Each month, we share summaries of recent Rare Diseases Clinical Research Network (RDCRN) grant-funded publications. Catch up on the latest RDCRN research below.
Jump to:
- Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)
- Frontiers in Congenital Disorders of Glycosylation Consortium (FCDGC)
- Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN)
- Lysosomal Disease Network (LDN)
- Myasthenia Gravis Rare Disease Network (MGNet)
- North American Mitochondrial Disease Consortium (NAMDC)
- Nephrotic Syndrome Study Network (NEPTUNE)
Listen to these summaries on the Rare Research Report podcast.
Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)
New Clinical Severity Index Helps Guide Management of Eosinophilic Esophagitis
For patients with eosinophilic esophagitis (EoE), an allergic inflammatory disease that damages the esophagus, therapeutic options and management are dictated by disease severity. However, the process for determining severity varies among practitioners. Reducing this variability could help improve clinicians’ ability to monitor EoE in an office setting.
In this study, researchers aimed to create an international consensus severity scoring index for EoE. First, a group of adult and pediatric EoE researchers and clinicians—as well as non-EoE allergy immunology and gastroenterology experts—reviewed existing literature on EoE in the context of progression and severity. Next, a steering committee reached consensus on important features of severity. These features were then distilled to categorize patients with EoE as having inactive, mild, moderate, or severe disease.
This new simplified scoring system, called the Index of Severity for Eosinophilic Esophagitis (I-SEE), can be completed at routine clinic visits. The system can help guide practitioners in EoE management by standardizing features of disease severity beyond eosinophil counts. To increase its utilization and functionality, authors note that I-SEE should be validated and refined using data from future clinical trials and routine clinical practice.
Dellon ES, Khoury P, Muir AB, Liacouras CA, Safroneeva E, Atkins D, Collins MH, Gonsalves N, Falk GW, Spergel JM, Hirano I, Chehade M, Schoepfer AM, Menard-Katcher C, Katzka DA, Bonis PA, Bredenoord AJ, Geng B, Jensen ET, Pesek RD, Feuerstadt P, Gupta SK, Lucendo AJ, Genta RM, Hiremath G, McGowan EC, Moawad FJ, Peterson KA, Rothenberg ME, Straumann A, Furuta GT, Aceves SS. A Clinical Severity Index for Eosinophilic Esophagitis: Development, Consensus, and Future Directions. Gastroenterology. 2022 Jul;163(1):59-76. doi: 10.1053/j.gastro.2022.03.025. Epub 2022 May 20. PMID: 35606197; PMCID: PMC9233087.
Frontiers in Congenital Disorders of Glycosylation Consortium (FCDGC)
ALG8-congenital disorder of glycosylation (ALG8-CDG) is a rare, inherited disorder that affects multiple systems in the body. Patients with ALG8-CDG commonly present with decreased muscle tone, intestinal problems, and liver problems.
In this study, researchers describe seven new individuals with ALG8-CDG, bringing the total to 26 individuals reported in medical literature. The team diagnosed these patients based on biochemical and molecular testing, identifying nine novel variants in ALG8. The cohort also includes the two oldest patients reported to date.
This study expands the phenotype of ALG8-CDG to include stable intellectual disability, autism spectrum disorder, and other neuropsychiatric symptoms. Researchers also expand the clinical features in a variety of organ systems. To improve clinical management, authors suggest a comprehensive evaluation and monitoring strategy.
Albokhari D, Ng BG, Guberinic A, Daniel EJP, Engelhardt NM, Barone R, Fiumara A, Garavelli L, Trimarchi G, Wolfe L, Raymond KM, Morava E, He M, Freeze HH, Lam C, Edmondson AC. ALG8-CDG: Molecular and phenotypic expansion suggests clinical management guidelines. J Inherit Metab Dis. 2022 Jun 18. doi: 10.1002/jimd.12527. Epub ahead of print. PMID: 35716054.
Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN)
Identifying Patterns of Hematologic Abnormalities in Aicardi Goutières Syndrome
Aicardi Goutières syndrome (AGS) is an inherited disease that is associated with early onset neurologic disability and systemic inflammation. Cytopenias—conditions in which there are lower-than-normal numbers of blood cells—are a potentially serious, but poorly understood, complication of AGS. As new treatment options are developed, it is important to understand the roles of the disease versus the treatment in hematologic abnormalities, allowing for better management of cytopenia.
In this study, researchers identified novel patterns of hematologic abnormalities in AGS. The team collected laboratory data throughout the lifespan from 142 individuals with AGS. Results showed that AGS results in multilineage cytopenias not limited to the neonatal period. Neutropenia, anemia, and thrombocytopenia were common. For patients on the treatment baricitinib, moderate to severe graded events of neutropenia, anemia, and leukopenia were more common, but rarely of clinical consequence.
Based on these results, authors recommend careful monitoring of hematologic parameters in children with AGS throughout the lifespan, especially while on therapy. Authors also note that AGS should be considered in children with neurologic impairment of unclear cause and hematologic abnormalities.
Adang LA, Gavazzi F, D'Aiello R, Isaacs D, Bronner N, Arici ZS, Flores Z, Jan A, Scher C, Sherbini O, Behrens EM, Goldbach-Mansky R, Olson TS, Lambert MP, Sullivan KE, Teachey DT, Witmer C, Vanderver A, Shults J. Hematologic abnormalities in Aicardi Goutières Syndrome. Mol Genet Metab. 2022 Aug;136(4):324-329. doi: 10.1016/j.ymgme.2022.06.003. Epub 2022 Jun 16. PMID: 35786528; PMCID: PMC9357135.
Lysosomal Disease Network (LDN)
New Method Reveals Insights on Podocyte GL3 Accumulation in Female Patients with Fabry Disease
Fabry disease is a lysosomal storage disorder caused by an abnormal enzyme that cannot break down some of the glycosphingolipids (cell membrane components containing fats with sugar molecules attached). The increase in one of these glycosphingolipids, called globotriasylceramide (GL3), is the hallmark of the disease and associated with cellular injury.
The Fabry disease gene is located on the X chromosome. In contrast to males, females have two X chromosomes in their cells, one of which is randomly inactivated. Although female patients can experience serious complications of Fabry disease, most studies focus on male patients in order to avoid confusion resulting from mosaicism caused by X chromosome inactivation.
In this study, researchers developed a new, unbiased method for GL3 estimation in podocytes (kidney cells that wrap around capillaries in the kidney filters called glomeruli) independent of mosaicism in female patients with Fabry disease. Researchers used this method to make age-matched comparisons between female and male patients and controls. Results showed that GL3 accumulation in podocytes that carry the gene defect in female patients with Fabry disease increases with age—and to the same levels as podocytes in males, all of which carry this defect. This accumulation is also associated with podocyte loss and leaking protein in the urine, predictors of kidney failure.
These studies indicate that Fabry-affected podocytes in female patients do not benefit from the circulating normal enzyme from their normal cells or from normal neighbor podocytes. Authors note that these novel findings help us better understand the mechanisms involved in Fabry disease complications and have important clinical implications.
Najafian B, Silvestroni A, Sokolovskiy A, Tøndel C, Svarstad E, Obrisca B, Ismail G, Holida MD, Mauer M. A novel unbiased method reveals progressive podocyte globotriaosylceramide accumulation and loss with age in females with Fabry disease. Kidney Int. 2022 Jul;102(1):173-182. doi: 10.1016/j.kint.2022.03.023. Epub 2022 Apr 26. PMID: 35483528; PMCID: PMC9233139.
Myasthenia Gravis Rare Disease Network (MGNet)
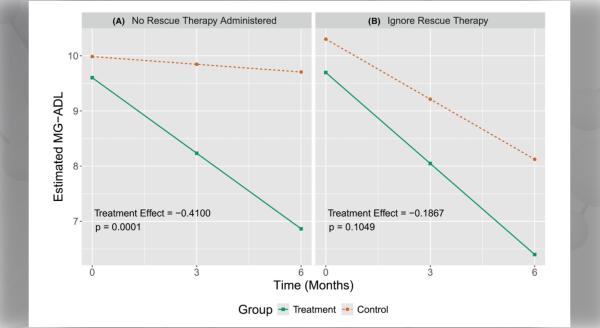
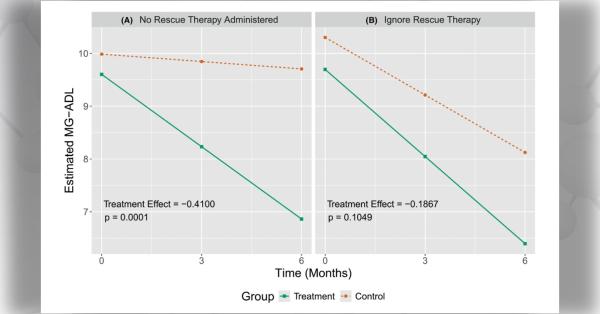
Myasthenia gravis (MG) is a neuromuscular disorder caused by an autoimmune response which blocks or damages acetylcholine receptors in muscles. Clinical assays—laboratory tests used to diagnose and monitor patients—only measure autoantibody binding. Therefore, these tests often provide limited insight on disease burden and therapeutic response.
To address these limitations, Dr. Kevin C. O’Connor and colleagues at Yale University developed a new assay for evaluating acetylcholine receptor autoantibody–mediated complement activity. Results suggested that a subset of patients lacks association between membrane attack complex formation and autoantibody binding or disease burden.
Authors note that this assay provides a better understanding of autoantibody mechanisms and may improve predictions for treatment response. Ultimately, these measurements could help assess disease progression and provide more individualized treatment plans.
Obaid AH, Zografou C, Vadysirisack DD, Munro-Sheldon B, Fichtner ML, Roy B, Philbrick WM, Bennett JL, Nowak RJ, O'Connor KC. Heterogeneity of Acetylcholine Receptor Autoantibody-Mediated Complement Activity in Patients With Myasthenia Gravis. Neurol Neuroimmunol Neuroinflamm. 2022 Apr 26;9(4):e1169. doi: 10.1212/NXI.0000000000001169. Erratum in: Neurol Neuroimmunol Neuroinflamm. 2022 Aug 3;9(5): PMID: 35473886; PMCID: PMC9128035.
North American Mitochondrial Disease Consortium (NAMDC)
Mitochondrial diseases are chronic, often inherited, multisystemic, genetic disorders involving dysfunction of the mitochondria (specialized cell structures that produce energy). The clinical, biochemical, and genetic characteristics of these diseases are diverse. Therefore, mitochondrial diseases are challenging to classify and diagnose.
To harmonize terminology, members of the North American Mitochondrial Disease Consortium (NAMDC) propose revised criteria for the clinical definition of mitochondrial disorders. First, they established a Diagnostic Criteria Committee with clinicians, researchers, diagnostic laboratory directors, statisticians, and data managers. Next, the Committee conducted a comprehensive literature review, an evaluation of current clinical practices and diagnostic modalities, surveys, and teleconferences.
After refining these criteria, the Committee reached a consensus on syndrome definitions for mitochondrial diseases. Authors note that updated criteria will standardize the diagnosis and categorization of mitochondrial diseases. These criteria can also help facilitate future natural history studies and clinical trials.
Emmanuele V, Ganesh J, Vladutiu G, Haas R, Kerr D, Saneto RP, Cohen BH, Van Hove JLK, Scaglia F, Hoppel C, Rosales XQ, Barca E, Buchsbaum R, Thompson JL, DiMauro S, Hirano M; North American Mitochondrial Disease Consortium (NAMDC). Time to harmonize mitochondrial syndrome nomenclature and classification: A consensus from the North American Mitochondrial Disease Consortium (NAMDC). Mol Genet Metab. 2022 Jun;136(2):125-131. doi: 10.1016/j.ymgme.2022.05.001. Epub 2022 May 13. PMID: 35606253; PMCID: PMC9341219.
Mitochondrial DNA depletion/deletions syndromes (MDDS) are a group of inherited disorders in which copies of the DNA (genetic material) within mitochondria (specialized cell structures that produce energy) are severely reduced in number. MDDS are frequently caused by defects in nucleoside and nucleotide metabolism, which is critical for mitochondrial DNA replication.
In this study, researchers report five individuals from four families who presented with ptosis (eyelid drooping), ophthalmoplegia (eye muscle weakness), other clinical manifestations, and multiple mitochondrial DNA deletions in muscle. The team identified three variants in the gene RRM1, which affect ribonucleotide reductase protein structure and function, leading to impairment of de novo nucleotide synthesis.
These results reveal that both recessive and dominant RRM1 variants cause MDDS. Additionally, these findings demonstrate that elements of the de novo nucleotide synthesis pathway may lead to MDDS.
Shintaku J, Pernice WM, Eyaid W, Gc JB, Brown ZP, Juanola-Falgarona M, Torres-Torronteras J, Sommerville EW, Hellebrekers DM, Blakely EL, Donaldson A, van de Laar I, Leu CS, Marti R, Frank J, Tanji K, Koolen DA, Rodenburg RJ, Chinnery PF, Smeets HJM, Gorman GS, Bonnen PE, Taylor RW, Hirano M. RRM1 variants cause a mitochondrial DNA maintenance disorder via impaired de novo nucleotide synthesis. J Clin Invest. 2022 Jul 1;132(13):e145660. doi: 10.1172/JCI145660. PMID: 35617047; PMCID: PMC9246377.
Nephrotic Syndrome Study Network (NEPTUNE)
Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are kidney disorders that damage the glomeruli, which are tiny blood vessels in the kidneys. Historically, classification of these disorders has been based on limited features of the glomeruli. Because disease course and treatment response are diverse among patients, a more detailed evaluation of kidney tissue features is needed.
In this study, researchers aimed to identify the most important descriptors of clinical outcomes in the Nephrotic Syndrome Study Network (NEPTUNE). The team used supervised machine learning methods—with pathology data collected through applying the NEPTUNE Digital Pathology Scoring System to NEPTUNE kidney biopsies—to evaluate predictors of disease progression, complete proteinuria remission, and treatment response.
Researchers found that the most predictive descriptors of outcomes included conventional and novel histologic and ultrastructural features from both glomerular and tubulointerstitial renal compartments. Authors note that standardized reporting of these descriptors could help inform predictions for clinical outcomes.
Zee J, Liu Q, Smith AR, Hodgin JB, Rosenberg A, Gillespie BW, Holzman LB, Barisoni L, Mariani LH; Nephrotic Syndrome Study Network (NEPTUNE); NEPTUNE Members. Kidney Biopsy Features Most Predictive of Clinical Outcomes in the Spectrum of Minimal Change Disease and Focal Segmental Glomerulosclerosis. J Am Soc Nephrol. 2022 Jul;33(7):1411-1426. doi: 10.1681/ASN.2021101396. Epub 2022 May 17. PMID: 35581011; PMCID: PMC9257823.
The Rare Diseases Clinical Research Network (RDCRN) is funded by the National Institutes of Health (NIH) and led by the National Center for Advancing Translational Sciences (NCATS) through its Division of Rare Diseases Research Innovation (DRDRI). Now in its fourth five-year funding cycle, RDCRN is a partnership with funding and programmatic support provided by Institutes, Centers, and Offices across NIH, including the National Institute of Neurological Disorders and Stroke, the National Institute of Allergy and Infectious Diseases, the National Institute of Diabetes and Digestive and Kidney Diseases, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Heart, Lung, and Blood Institute, the National Institute of Dental and Craniofacial Research, the National Institute of Mental Health, and the Office of Dietary Supplements.